物构知识点(正在学习中)
不断更新中
由于KaTeX无法直接渲染化学式,故直接采用Typora渲染,可能导致目录等功能暂缺。
原子结构与性质
研究历史
| 主体 | 道尔顿 | 汤姆生 | 卢瑟福 | 玻尔 | 量子力学 |
|---|---|---|---|---|---|
| 年代 | 1803 | 1904 | 1911 | 1913 | 1926 |
| 依据 | 元素化合时的质量比例关系 | 发现电子 | α粒子散射 | 氢原子光谱 | 近代科学实验 |
| 主要内容 | 原子是不可再分的实心球体 | 葡萄干布丁模型 | 行星模型 | 轨道式原子模型 | 电子云模型 |
基态原子的核外电子排布
电子云
- 用小点的疏密来描述电子在该区域内出现的机会的大小
排布规律
能量最低原理:原子核外电子总是先占有能量最低的电子轨道。
泡利不相容原理:每个原子轨道上最多只能容纳2个自旋方向相反的电子
- 电子空间运动状态不同:三不同:能层-能级-轨道伸展方向(即轨道数)
- 电子运动状态不同:空间运动状态+自旋 即每一个电子的运动状态都不同
洪特规则:当电子排布在同一能级的不同轨道时,基态原子中的电子总是优先单独占据一个轨道,而且自旋状态相同
未成对电子
空轨道数 未成对电子数 成对电子数 1 2 1 2 1 2 p p2 p1 p1/5 p2/4 p4 p5 d d4 d3 d1/9 d2/8 d6 d7 未成对电子数 所在族及典型元素 0 ⅡA、ⅡB、0族 1 ⅠA、ⅠB、ⅢB、ⅢA、ⅦA 2 ⅣA、ⅥA、ⅣB、Ⅷ 3 ⅤA、ⅤB、Ⅷ 4 Ⅷ(Fe) 5 ⅦB(Mn) 6 ⅥB(Cr)
当轨道位于全满或半满时,比较稳定
Cr:[Ar]3d54s1
Cu:[ar]3d104s1
解释某些离子的稳定性问题
Fe2+:[Ar]3d6
Fe3+:[Ar]3d5
Fe3+的3d轨道处于半满状态,更稳定。
基态原子和激发态原子
- 基态原子:能量处于最低状态的原子。
- 激发态原子:能量处于较高状态的原子。
- 基态原子(1s22s22p3)→激发态原子(1s22s12p4):需要吸收能量,会形成吸收光谱。
- 激发态原子(1s22s12p4)→基态原子(1s22s22p3):会以可见光的形式放出能量,会形成发射光谱。焰色反应原理。
四种表示方法
- 电子排布式
例: Cr:1s22s22p63s23p63d54s1
- 简化表示式
例:Cu:[Ar]3d104s1
- 价电子排布式
例:Fe:3d64s2
- 电子排布图/轨道排布式
例:
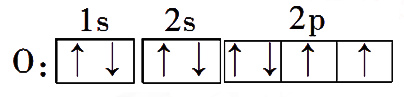
原子轨道
- 电子层
| 由内至外电子层顺序 | 电子层符号 | 亚电子层数目 | 原子轨道数目n2 | 可容纳电子数目2n2 |
|---|---|---|---|---|
| 1 | K | 1 | 1 | 2 |
| 2 | L | 2 | 4 | 8 |
| 3 | M | 3 | 9 | 18 |
| 4 | N | 4 | 16 | 32 |
- 原子轨道
常把电子出现的概率大于90%的空间圈出来,并将这种电子云轮廓图称为原子轨道。
| 由内至外顺序 | 名称 | 形状 | 伸展方向 | 可容纳电子数目 |
|---|---|---|---|---|
| 1 | s | 球形 | 1 | 2 |
| 2 | p | 纺锤形/哑铃型 | 3 | 6 |
| 3 | d | 梅花型 | 5 | 10 |
| 4 | f | 复杂 | 7 | 14 |
能量比较
不同能层,同一能级,能层序数越大,能量越高
例:1s<2s<3s<4s
同一能层,不同能级,ns<np<nd<nf
相同能层,相同能级,不同轨道,能量大小相等
例:3Px=3Py=3Pz
不同能层,不同能级——能级交错现象
公式:ns<(n-2)f<(n-1)d<np

价电子数
- 主族元素(A)和0 族:价电子= 外围电子= 最外层电子= 主族序数 (ns1-2或ns2np1-6 )
- 副族元素(B)和VIII 族:价电子= 外围电子= 副族序数 ( (n-1)d1-10ns1-2 )
红色部分相加为列数
元素第一电离能和电负性及其变化规律
电负性: 衡量元素在化合物中吸引电子的能力。
氟的电负性为4.0
变化规律:
- 同周期(从左到右):依次增大
- 同主族(自上而下):依次减小
一般来说,金属元素的电负性小于非金属元素的电负性
电负性化合价
化合价/电负性结构
第一电离能: 某元素的气态原子失去一个电子形成+1价气态阳离子所需的最低能量,用符号I1表示
变化规律:
- 同周期(从左到右):增大趋势(注意ⅡA、ⅤA的特殊性)
- 同主族(自上而下):依次减小
例:O原子半径比C原子半径小,且核电荷数比C的大,故O原子对外层电子吸引力更强,更难失电子,故其I1大于C
同周期元素,ⅡA族(np0)和ⅤA族(ns2np3),因p轨道处于全空或半满状态,比较稳定,所以其第一电离能大于同周期右侧相邻的ⅢA族、ⅥA族元素,如第一电离能Mg>Al,P>S。

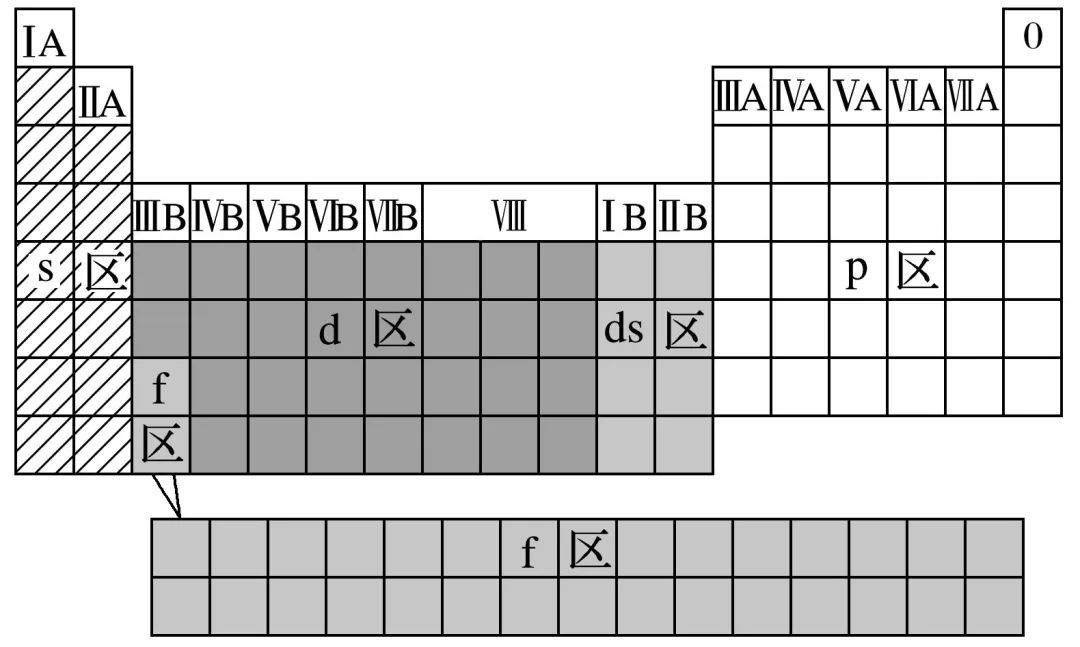
元素周期表中区的划分
标准:价电子排布
| 区 | 族 | 最外层电子 |
|---|---|---|
| s区 | ⅠA、ⅡA两族元素 | ns1-2 |
| p区 | ⅢA-ⅦA族元素及0族元素 | ns2np1-6 |
| d区 | ⅢB-ⅦB,Ⅷ族 | (n-1)d1-8ns2 |
| ds区 | ⅠB、ⅡB族 | (n-1)d10ns1-2 |
| f区 | 镧系、锕系元素 |
晶体结构与性质
分类
| 类别 | 粒子间作用力 | 特征 |
|---|---|---|
| 分子晶体 | 分子间作用力(范德华力、氢键) | 低熔沸点 |
| 离子晶体 | 离子键 | 较高熔沸点,在水溶液/熔融能导电 |
| 原子晶体 | 共价键 | 高熔沸点 |
| 金属晶体 | 金属键 | 导电、传热、延展性 |
金属晶体
金属键
- 金属阳离子和自由电子之间强烈的相互作用
- 存在于金属单质/合金/金属簇化合物中
- 金属键没有方向性——延展性
- 自由电子在外电场作用下定向移动——导电性
- 通过自由电子的运动把能量从温度高的地方向温度低的地方传递——导热性
- 金属离子半径越小,离子电荷数越多,金属键越强,金属熔、沸点就越高。
金属晶体堆积模型
密置层和非密置层
- 非密置层:每个圆球和其他四个球相切
- 密置层:每个圆球和其他六个球相切
(金属的)原子化热
- 1mol金属固体完全气化成相互远离的气态原子时吸收的能量
| 类别 | 例子 | 示意图 | 晶胞占有原子数 | 原子半径r与立方体边长a的关系 | 配位数 | 空间利用率 |
|---|---|---|---|---|---|---|
| 简单立方堆积 | 钋(Po) |  | 1 | a=2r | 6 | ≈52% |
| 体心立方堆积 | 钠、钾、铬、钼、钨 |  | 2 | =4r | 8 | ≈68% |
| 面心立方堆积 | 金、银、铜、铅 |  | 4 | =2r | 12 | ≈74% |
| 六方立方堆积 | 镁、锌、钛 |  | 2 | 12 | ≈74% |
- 关于面心和六方的一些图片
左面心,右六方

离子晶体
离子键
- 阴阳离子之间的强烈的相互作用
- 没有方向性和饱和性
- 含有离子键的化合物就是离子化合物
- 包括引力和斥力
晶格能U
- 拆开1mol离子晶体使之形成气态阳离子和气态阴离子时所需的能量。
- 晶格能越大,表明离子晶体中的离子键越强。
- 一般而言,晶格能越大,离子晶体的熔点越高,硬度越大。
- 影响因素:阴阳离子的电荷数越多,离子间的核间距越小,晶格能越大
离子配位数
- 一种离子周围紧邻的带相反电荷的离子数目。
- 离子晶体中离子的配位数主要取决于阴、阳离子的相对大小。
离子晶体
由阴阳离子通过离子键结合而成的晶体
构成微粒:阴离子和阳离子
判断方法
- 熔融状态下能导电的化合物
- 出现Na+,K+,Ba2+,Mg2+,NH4+六种离子的其中一种的一定是离子晶体
- 无任何信息下的含金属元素的(除AlCl3)
- 有阳离子和阴离子(只有阳离子:可能为金属晶体)
作用力判断
- 离子晶体中作用力一定有:离子键
- 离子晶体中作用力可能有:共价键
- 熔化时破坏的作用力:离子键
故可得:氯化钠溶液或熔融氯化钠中没有离子键 离子化合物熔化只断键,属于物理变化
物理性质(括号中为例外)
- 熔沸点较高(原子晶体)
- 1固体不导电,2熔融状态能导电,3水溶液一般导电(13例外:HCl)
- 大多易溶于水,难溶于有机溶剂
热稳定性
例:碳酸盐:X2++CO32-=XO+CO2
- 分解温度:MgCO3<CaCO3<SrCO3<BaCO3
- 原因:X-O键越强,越容易把O2-从CO32-中脱离出来
- 写法:碳酸盐分解本质为CO32-生成CO2和O2-,Mg2半径最小,与O2-结合能力最强,故MgCO3最易分解
常见离子晶体
| 类型 | NaCl | CsCl | ZnS | CaF2 |
|---|---|---|---|---|
| 示意图 |  | 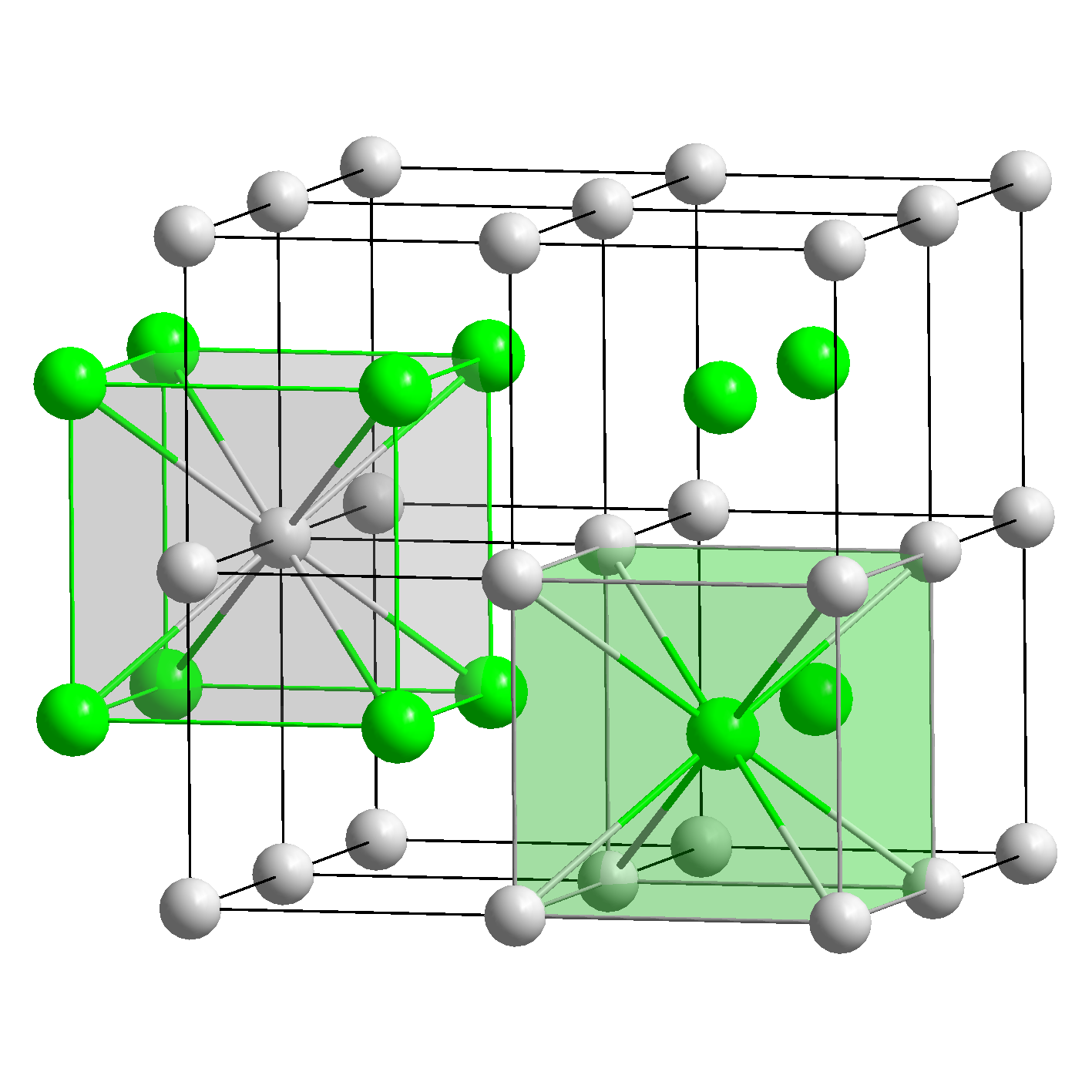 |  | 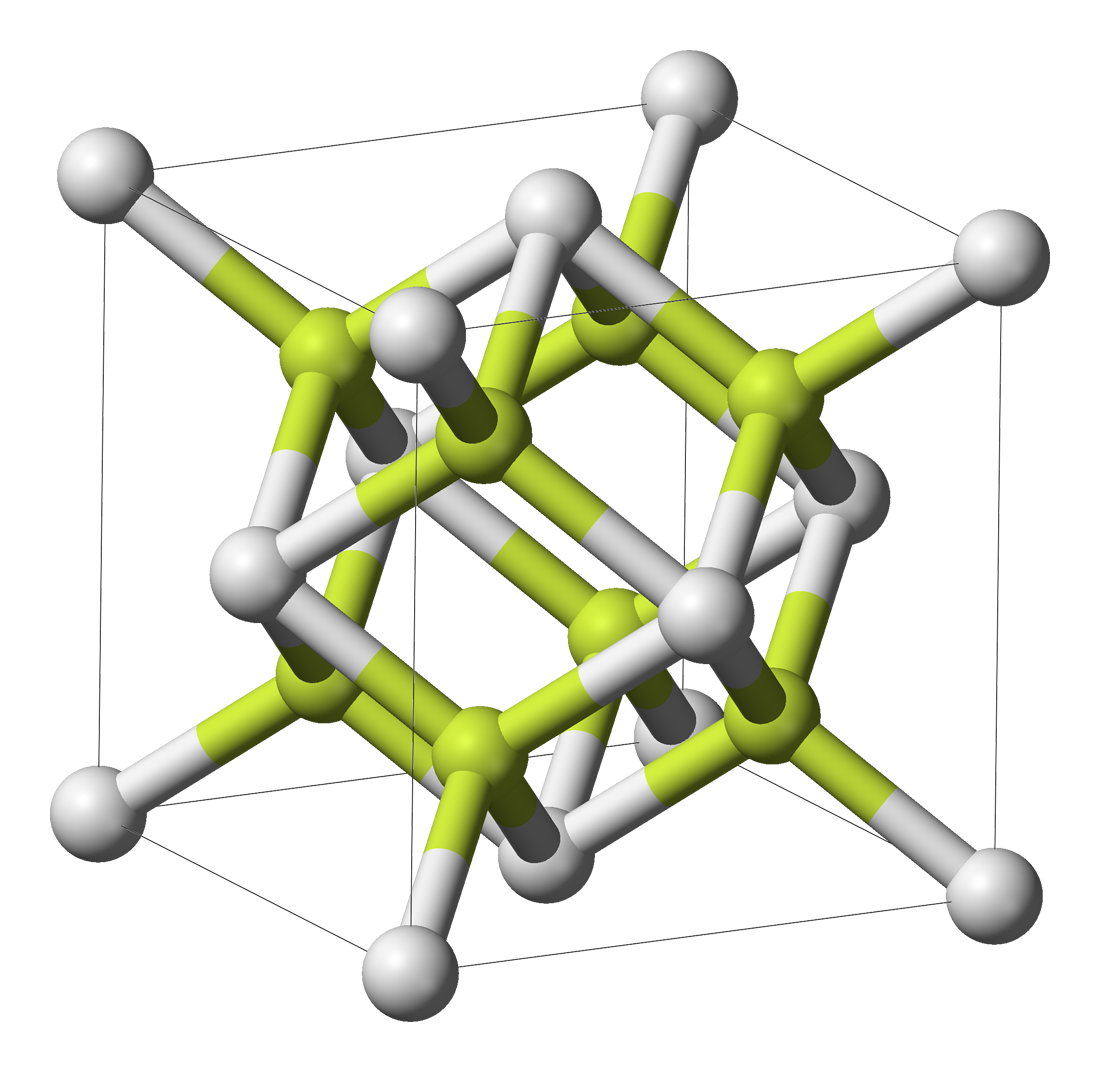 |
| 堆积方式 | 面心立方堆积 | 体心立方堆积 | 类金刚石堆积 | 面心立方堆积 |
| 离子数目 | 4个Na+和4个Cl- | 1个Cs+和1个Cl- | 4个Zn2+和4个S2- | 4个Ca2+和8个F- |
| 离子配位数 | Na+的配位数为6,Cl-的配位数为6 | Cs+的配位数为8,Cl的配位数为8 | Zn2+的配位数是4,S2-的配位数是4 | Ca2+的配位数为8,F-的配位数为4 |
| 最近同离子数 | 在每个Na+周围最近且等距离的Na+有12个,在每个Cl-周围最近且等距离的Cl-有12个 | 在每个Cs+周围最近且等距离的Cs+有6个,在每个Cl-周围最近且等距离的Cl-有6个 | 在每个Zn2+周围最近且等距离的Zn2+有6个,在每个S2-周围最近且等距离的S2-有6个 | 在每个Ca2+周围最近且等距离的Ca2+有12个,在每个F-周围最近且等距离的F-有6个 |
| 配位数 | 6(在所有八面体空隙插入原子) | 4(在互不相邻的四面体空隙插入原子) | 4(在所有四面体空隙插入原子) | |
| 阴阳离子最近距离 | Zn2+与S2-的最近距离为a(体对角线的四等分点) | Ca2+与F-的最近距离为a | ||
| 空隙填充 | Zn2+形成8个四面体空隙,S2-填入其中4个四面体空隙,空隙填充率50% | Ca2+形成8个四面体空隙,F-填入其中8个四面体空隙,空隙填充率100% | ||
| 其他 | 离Na+周围最近的Cl-构成的空间构型为空间正八面体 |
原子晶体
共价键
- 原子间通过共用电子对形成的化学键
- 成键微粒:原子
- 共价键形成的本质:当成键原子相接近时,原子轨道发生重叠,自旋方向相反的未成对电子形成共用电子对。两原子核间的电子密度增加,体系的能量降低。
- 具有饱和性。
- 具有方向性。(s轨道和s轨道重叠形成的共价键无方向性)
共价键的参数
- 键能:拆开1mol共价键所吸收的能量 或 形成1mol共价键所释放的能量。
- 键长:两个成键原子的核间距离叫做该化学键的键长(约等于成键两原子半径之和)
- 键角:多原子分子中的两个共价键之间的夹角。
共价键的分类
形成共价键的原子轨道重叠方式
σ键:“头碰头”重叠
形成:沿键轴方向以“头碰头”的方式发生原子轨道(电子云)重叠
类别
- s-sσ键:由两个s原子轨道重叠形成的σ键(无方向性)
- s-pσ键:由一个s原子轨道和一个p原子轨道重叠形成的σ键
- p-pσ键:由两个p原子轨道重叠形成的σ键
特征
- 形成σ键的原子轨道重叠程度较大,故较稳定
- 以形成σ键的两原子核的连线为轴,任何一个原子均可以旋转,并不破坏σ键的结构
π键:“肩并肩”重叠
形成:两个原子的电子以“肩并肩”的方式发生原子轨道重叠。
示意图 
分类
- 定域π键:两原子间形成的π键
例:N2 2个π键
离域π键:多原子间形成的π键
表达方式:
- a:成键原子数(偶数,且≥4)
- b:成键电子数
例:(CO2,O3),,,(苯)
形成条件
- 这些原子多数处于同一平面上
- 这些原子有相互平行的p轨道
- p轨道上的电子总数小于p轨道数的两倍
特征
- 形成π键时电子云重叠程度比σ键小,故较不稳定,且键长较σ键短。
“键长越短,键能越大”只适用于两个键都是σ键的情况下
有机物中C可形成单键、双键、叁键,但S难以形成双键、叁键:S原子半径大,“肩并肩”重合形成轨道重叠低,难以形成π键。
- 以形成π键的两个原子核的连线为轴,任意一个原子并不能单独旋转。
形成共价键的电子对是否偏移
- 极性键 共用电子对发生偏移(组成共价键的两个原子不同)
- 非极性键 共用电子对不发生偏移(组成两个共价键的原子相同)
原子间共用电子对的数目
- 单键 原子间有一对共用电子对
- 双键 原子间有两对共用电子对
- 三键 原子间有三对共用电子对
成键电子的来源
普通共价键:成键的两个原子乙方提供具有单电子的轨道,另一方也提供具有单电子的轨道而形成的共价键
配位键:成键的两个原子或离子一方提供孤电子对,另一方提供空轨道而形成的共价键
形成条件:一个原子提供孤对电子。另一原子提供空轨道
表示方法:
强弱比较:电负性
例:NH3中N有孤对电子,可以形成配位键;NF3中N也有孤对电子,但无法形成配位键的原因:F电负性比N强,NF3中N周围电子云密度低,故NF3中N难以提供孤对电子,形成配位键。
- 粒子分类
提供空轨道:中心原子/离子:
- H+
- ⅢA: B、Al
- 过渡金属:Ag+、Zn2+、Cu2+、Fe2+、Fe3+等
提供孤对电子:配体
- ⅦA: F-、Cl-、Br-、I-
- ⅥA: H2O、OH-、OCN-
- ⅤA: NH3、N2H4、乙二胺()
- ⅣA: CO、CN-
八电子稳定结构:最外层电子数+共价键数目
特别提醒
- 单键为σ键;双键或三键,其中一个为σ键,其余为π键。
- 配位键为一种特殊的共价键,形成配位键的条件是成键原子一方(A)能够提供孤电子对,另一方(B)具有能够接受孤电子对的空轨道,可表示为A―→B。。
- s轨道形成的共价键全部是σ键;杂化轨道形成的共价键全部为σ键。
原子晶体
晶体中所有原子都是通过共价键结合的晶体
分子晶体
范德华力
把分子聚集在一起的作用力
特征:
- 无方向性、饱和性
- 分子间作用力(范德华力和氢键)比化学键弱(范德华力约比化学键键能小1-2个数量级。
判断:所有的分子晶体中都存在范德华力,随着三态变化,范德华力会相应变化。
影响因素:组成和结构相似时,M越大,范德华力越强,分子晶体的熔沸点越高。
主要影响物质的熔沸点。
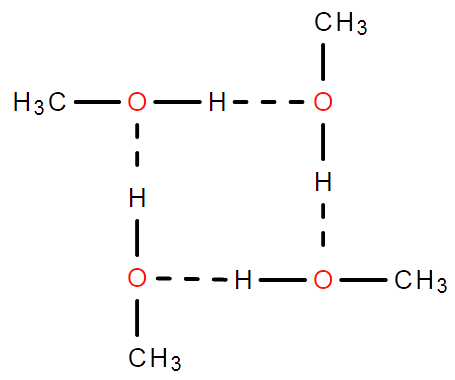
氢键
由已经与电负性很强且原子半径很小的原子形成共价键的氢原子与另一个分子中电负性很强原子半径很小且有孤对电子的原子之间的作用力。
表达方法:
:氢给予基
:氢接受基 (自身无法形成氢键,但可与外界分子形成氢键)
- 与均为N、O、F中的一种
- 必有孤对电子
- 必须为N、O、F且连在什么分子上不重要
特征
- 氢键不属于化学键,是一种分子间作用力,氢键键能很小,约为化学键的十分之几,但比范德华力强
- 氢键具有一定的方向性与饱和性
应用
存在
- 给予基、接受基: (自身分子间也可形成氢键)
- 给予基:
- 接受基:
熔沸点判断
分子间氢键>无氢键>分子内氢键
都为分子间氢键时
- 氢键数目:一般数目越多,熔沸点越高
- 氢键极性(键能):氢键极性越高,氢键键能越强
分子间氢键
水杨醛(邻羟基苯甲醛) 对羟基苯甲醛 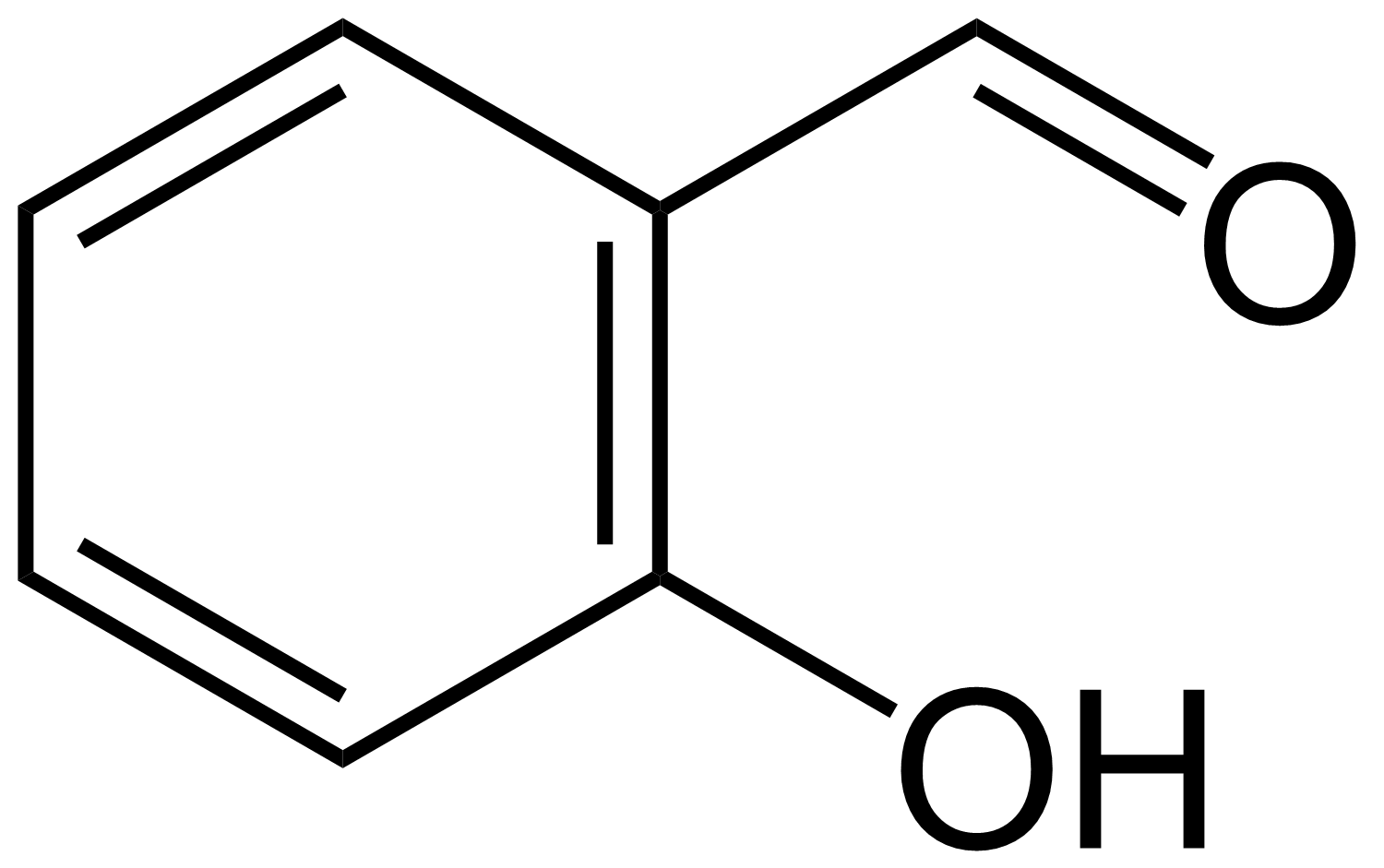

分子内部可形成氢键 分子内部不可形成氢键 硝酸 邻硝基苯酚 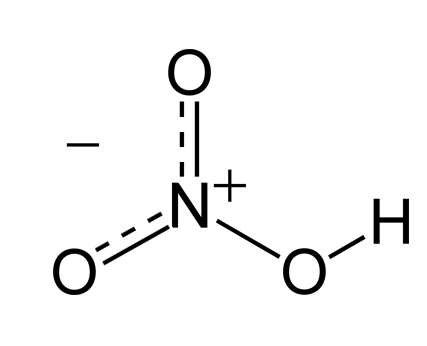

例:与相比,后者熔沸点更高的原因:后者与相连,可形成分子间氢键。
“缔合问题”
问:分子经常写成/相对分子质量测量值比理论值大的原因
答:分子间存在氢键。形成缔合分子。
二聚甲酸 四聚甲醇 
晶体小结
| 离子键 | 共价键 | 金属键 | |
|---|---|---|---|
| 定义 | 阴阳离子之间的强烈的相互作用 | 原子间通过共用电子对形成的化学键 | 金属阳离子和自由电子之间强烈的相互作用 |
| 微粒 | 阴离子和阳离子 | 原子 | 金属阳离子和电子 |
| 本质 | 当阴阳离子之间的静电引力和静电斥力达到平衡时,阴阳离子保持一定的平衡核间距,整个体系达到能量最低状态。 | 当成键原子相接近时,原子轨道发生重叠,自旋方向相反的未成对电子形成共用电子对。两原子核间的电子密度增加,体系的能量降低。 | 金属原子的部分或全部外围电子受原子核的束缚比较弱,在金属晶体内部可以从原子上“脱落”下来,形成自由流动的电子。失去部分或全部外围电子形成的金属离子与自由电子间存在强烈相互作用。 |
| 特征 | 没有方向性、饱和性 | 有方向性、饱和性 | 没有方向性、饱和性 |
| 强弱判断 | 阴阳离子的电荷数越多,离子间的核间距越小,晶格能越大,离子键越强 | 键长、类型 | 金属离子半径越小,离子电荷数越多,金属键越强 |
| 影响性质 | 离子晶体熔沸点 | 分子晶体熔沸点,分子稳定性 | 金属熔沸点,延展性,导电性,导热性 |
立方晶胞各物理量的计算
a3×ρ×NA=n×M
- a:晶胞棱长
- ρ:密度
- NA:阿伏加德罗常数的值
- n:1mol晶胞中所含晶体的物质的量
- M:晶体的摩尔质量
熔沸点比较规律
不同类型晶体的熔、沸点高低一般规律
- 原子晶体>离子晶体>分子晶体,
- 金属晶体的熔、沸点差别很大,如钨、铂等熔点很高,汞、铯等熔点很低。
同类晶体:
- 原子晶体:看成键原子半径和,半径和越小,晶体的熔、沸点越高、硬度越大。
- 离子晶体:依据静电原理进行分析,一般地说,阴、阳离子的电荷数越多,离子半径越小,则离子键越强,相应地晶格能越大,熔、沸点越高。
- 分子晶体:组成和结构相似的物质,相对分子质量越大,熔沸点越高(若分子间存在氢键,则含有氢键的熔、沸点高些)。
组成和结构不相似的分子晶体(相对分子质量接近),分子的极性越大,其熔、沸点越高。如CO>N2,CH3OH>CH3CH3。 - 金属晶体:金属离子半径越小,离子电荷数越多,金属键越强,金属熔、 沸点就越高。如熔、沸点:Al>Mg>Na。
分子空间结构和物质性质
杂化和空间构型
杂化轨道理论
概念:原子在形成分子时,为增强成键能力,同一个原子中能量相近(s、p、d)的几个原子轨道可以相互叠加进行重新组合,形成能量、形状和方向与原轨道不同的新的原子轨道。这种原子轨道重新组合的过程称为原子轨道的杂化,所形成的新的原子轨道称为杂化轨道
注意
- 只有在形成分子的过程中,中心原子能量相同,原子轨道才能进行杂化。孤立的原子不可能发生杂化。
- 只有能量相近的轨道才能互相杂化。
- 杂化前后,原子轨道的总数不变。
- 杂化轨道只用于放形成σ键的电子或孤电子对,不能放形成π键的电子。
最常见的杂化轨道类型
基本类型 sp sp2 sp3 杂化轨道数目 2 3 4 杂化轨道间的夹角 180° 120° 109.5° 示意图 


杂化轨道的空间构型 直线 平面正三角形 空间正四面体 分子的空间构型 直线型 V型(2个配位原子) 例:SO2
平面正三角型(3个配位原子) 例:BF3V型(2个配位原子) 例:H2O
三角锥结构(3个配位原子) 例:NH3
四面体结构(4个配位原子)另附
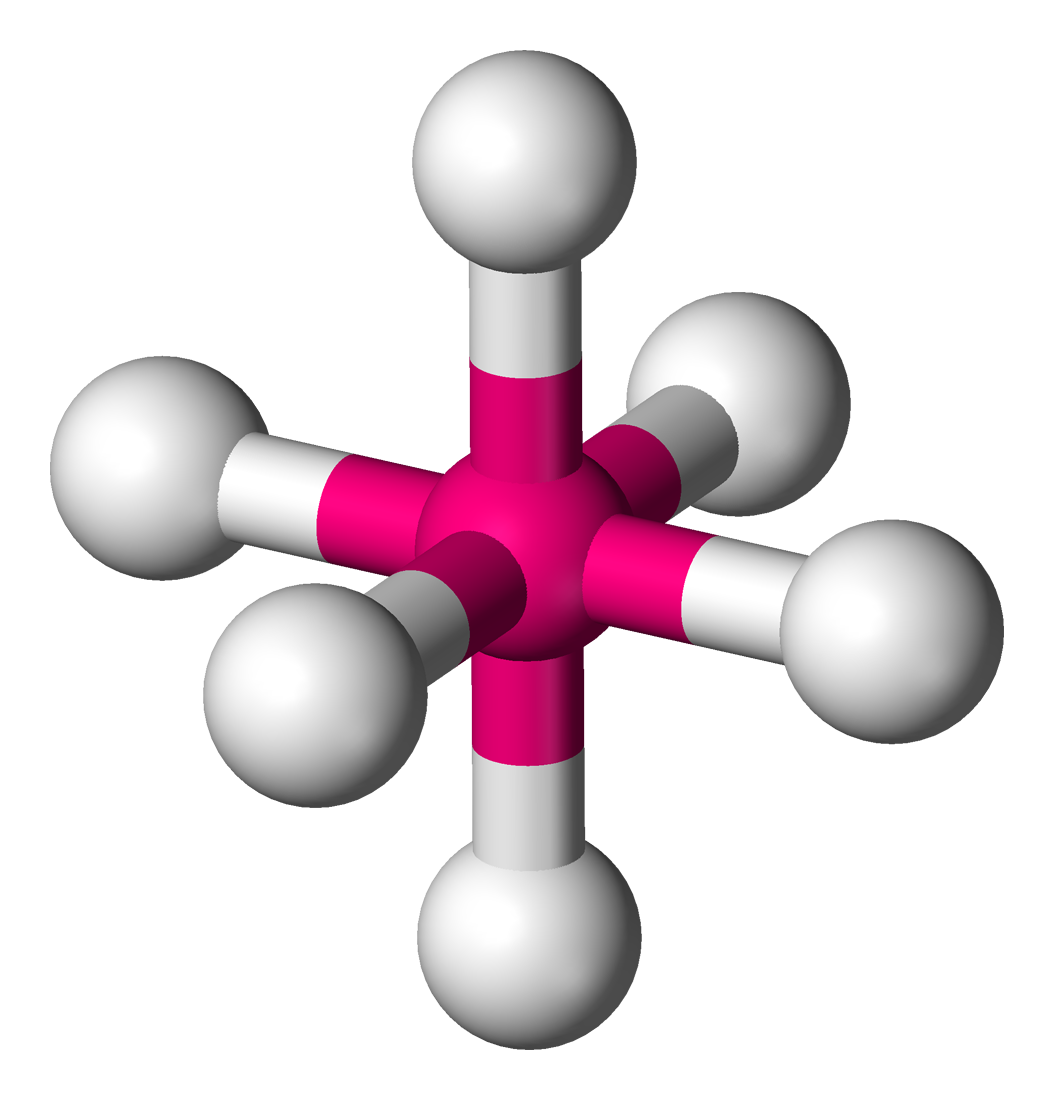
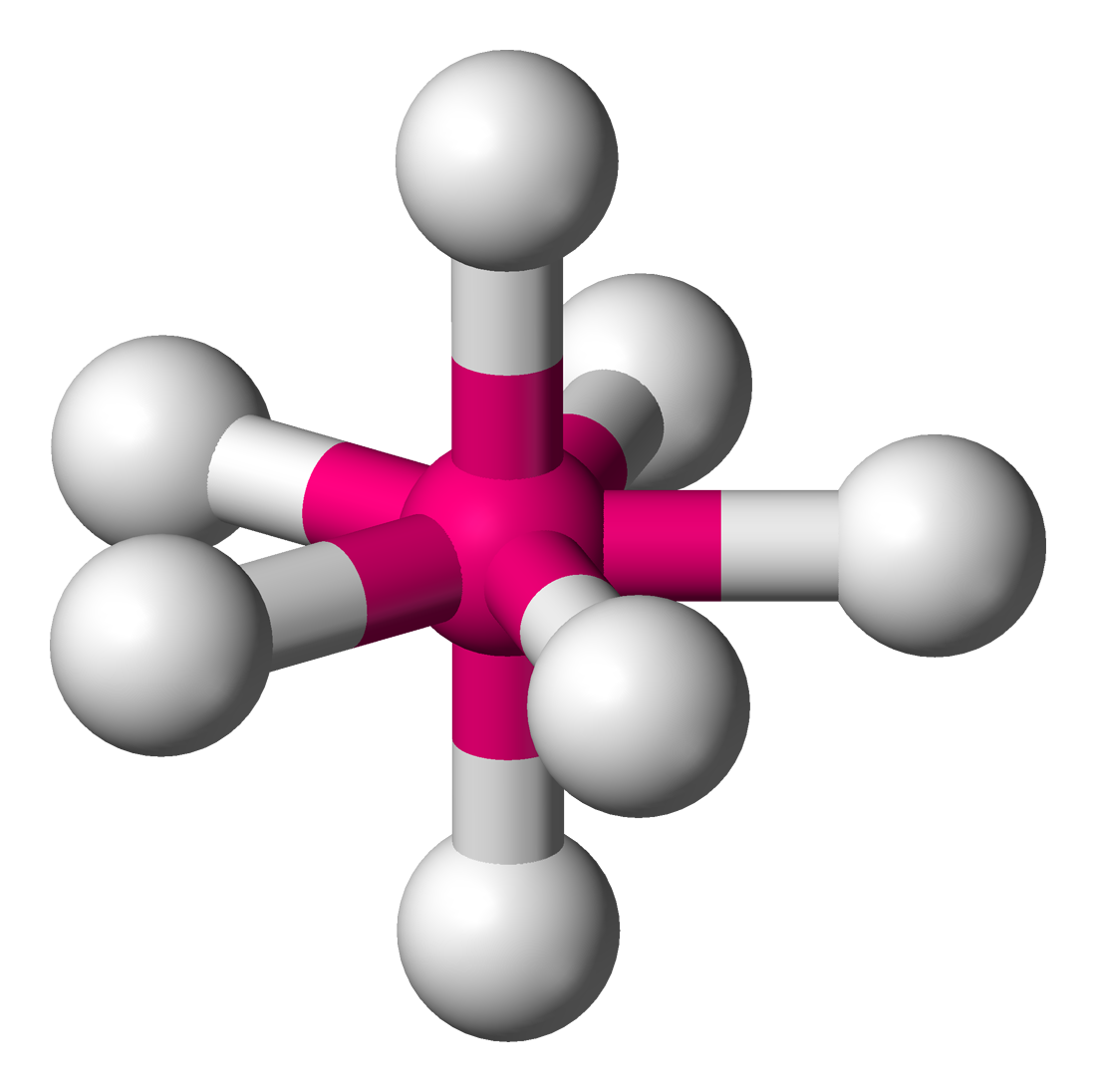
sp3d sp3d2 sp3d3 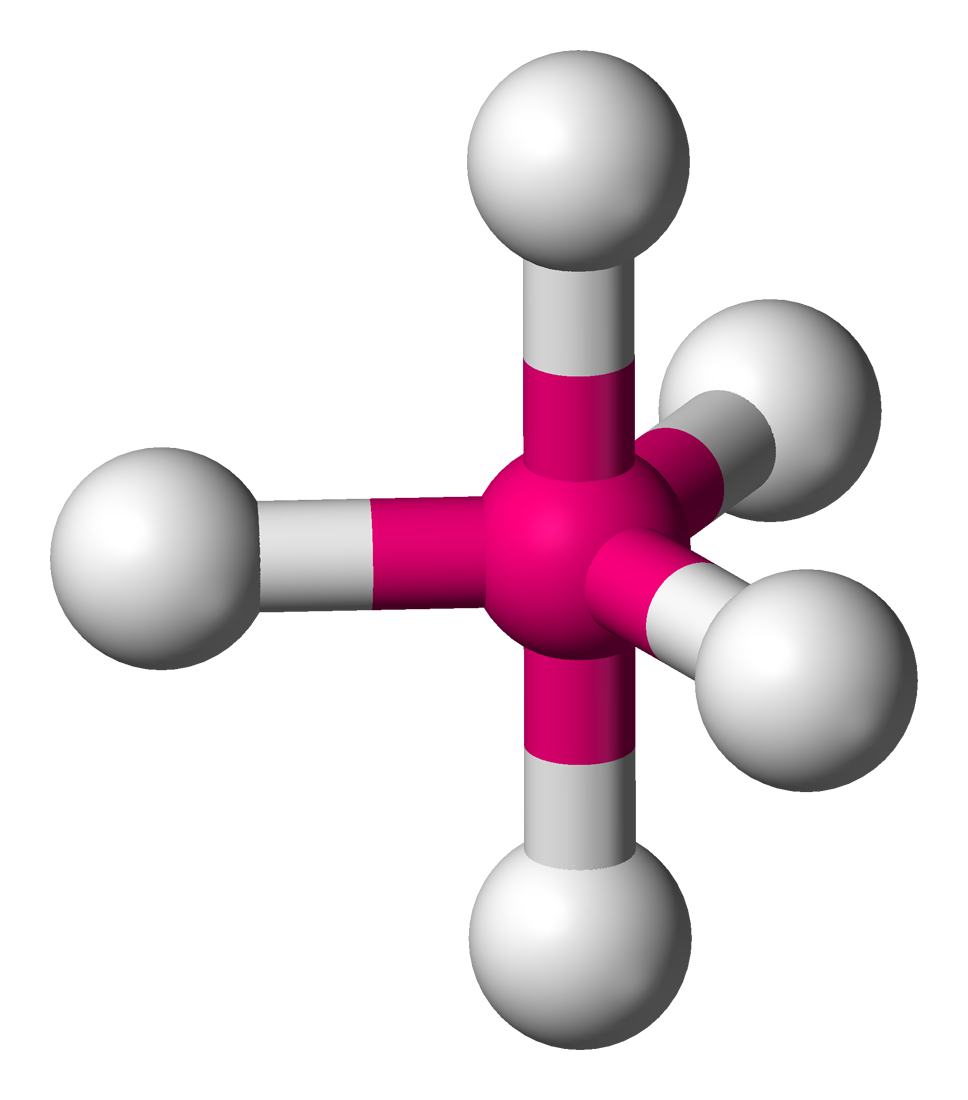
杂化轨道数目的具体算法
价层电子对互斥理论
- 中心原子的价电子对数=中心原子的杂化轨道数目=中心原子周围的σ键键数 +中心原子的孤电子对数。
- 中心原子周围的σ键键数=(中心原子价电子数 + 配位电子数)÷2
- 中心原子的孤电子对数=中心原子价电子数 - 中心原子成键数目
- 判断杂化轨道数目的步骤:配位原子数、孤对电子数→杂化轨道数或价层电子对数→杂化类型
碳原子杂化类型
- 周围都是单键→sp3
- 周围出现一个双键→sp2
- 周围出现一个叁键→sp
利用配位原子数推杂化类型
- 配位原子数为4→sp3
- 配位原子数为3→sp3/sp2
- 配位原子数为2→sp3/sp2/sp
利用空间构型推杂化类型
- 四面体形→sp3
- 三角锥形→sp3
- 平面三角形→sp2
- 直线形→sp
- V形→sp/sp3
- 八面体形→sp3d
- 三角双锥→sp3d
分子构型的键角
孤对电子对孤对电子的排斥力>孤对电子对成键电子的排斥力>成键电子对成键电子的排斥力
判断杂化类型,若不同
sp>sp2>sp3
看孤对电子数
孤对电子数越多,则键角越小。
看中心原子或配位原子电负性
中心原子电负性越大,键角越大;配位原子电负性越大,键角越小。
等电子原理
- 等电子体:价电子总数和原子总数相同的分子、离子或基团,其电子总数不一定相等。
- 等电子原理:等电子体具有相同的结构特征,性质也相近。
方法:同族替换;族数替换;电荷转换
常见等电子体
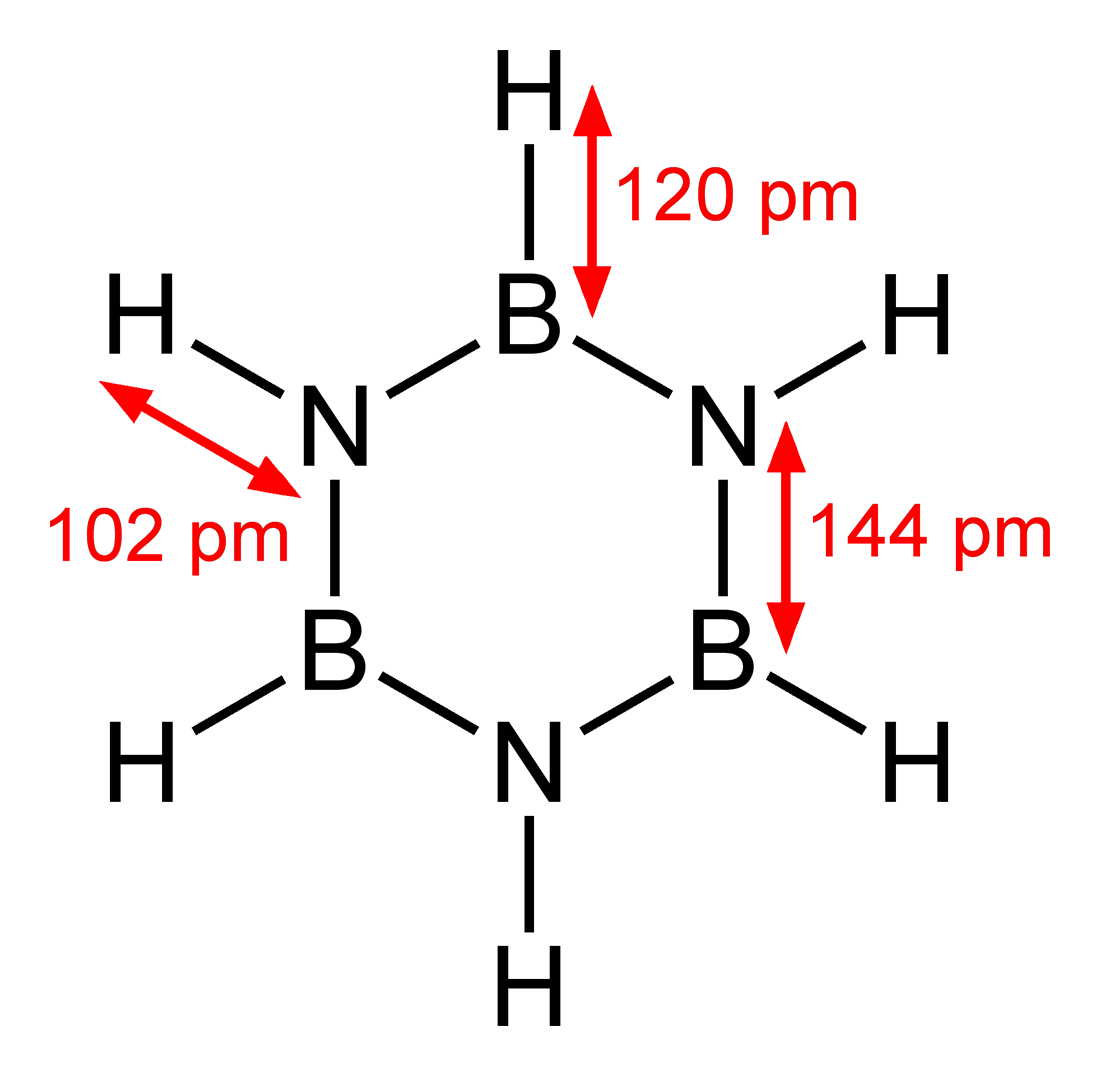
(苯)与(环硼氮烷)
苯 环硼氮烷 
分子的极性
概念
- 极性分子:正、负电荷中心不重合
- 非极性分子:正、负电荷中心重合
判断
双原子分子:看键的极性
极性键极性分子
非极性键非极性分子
多原子分子:键的极性+空间构型(键长、键角)
ABm型分子的判断方法
- 中心原子A的化合价的绝对值=A原子的最外层电子数非极性
- 中心原子A的化合价的绝对值≠A原子的最外层电子数极性
应用:相似相溶
手性分子
- 手性碳原子:连接四个不同的原子或基团的碳原子
配合物的形成和应用
定义:
组成
中心原子:提供空轨道接受孤电子对的离子或原子(大多数为过渡系列)
配位体:提供孤电子对的分子或离子
配位原子:配位体中提供孤电子对的原子
配位数:实际与中心原子以配位键结合的原子个数
中心原子电荷数=配离子电荷数-配体电荷总数
:中心原子
:配位体
:配位原子
:配位数
:内界
:外界
配合物的外界和内界在水中完全电离,内界很稳定
部分配合物只有内界,如
结构
化学键
内界的空间构型
配位数 2 4 4 6 杂化类型 sp sp3 dsp2 sp3d 空间构型 直线形 四面体形 正方形 八面体形 实例 顺反异构
存在于某些双键化合物或环状化合物中的一种立体异构现象。
顺-2-丁烯 反-2-丁烯 

性质
- 颜色:多数配合物具有颜色
- 溶解度:大部分配合物易溶于水
- 稳定性:配位键越强,配合物越稳定